


Home » Course Layouts » Free Course Layout Udemy
MSE 597G An Introduction to Molecular Dynamics
By Alejandro Strachan
Purdue University
0
1
English
English [CC]
- Learn basic syntax that can apply to any language.
- Learn what is a programming language and the basic concepts for beginners.
- Understand what is Javascript in it's truest form.
- Know the basic syntax of Javascript.
- Know some hidden quirks in Javascript.
Description
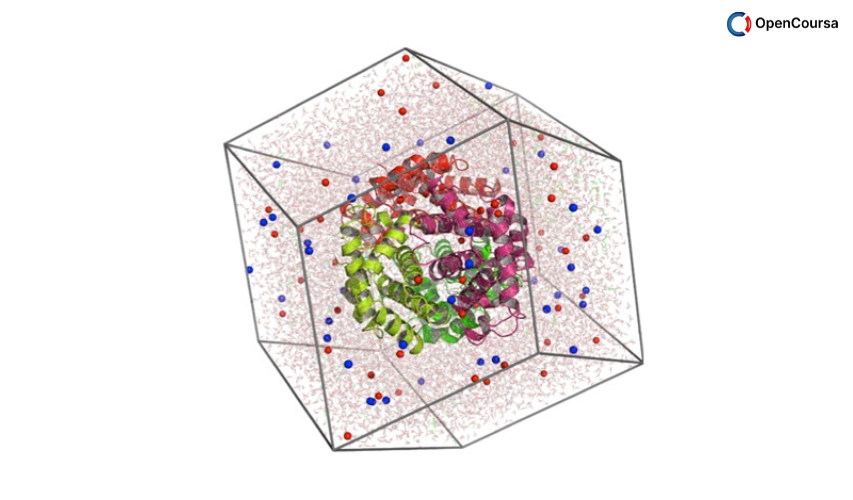
Molecular dynamics (MD) simulations are playing an increasingly important role in many areas of science and engineering, from biology and pharmacy to nanoelectronics and structural materials. Recent breakthroughs in methodologies and in the development of first principles-based interatomic potentials significantly increased the range of applicability of MD and the accuracy of its predictions even for new materials not yet fabricated or synthesized. Such predictive power indicates that MD has the potential to play a key role in guiding the design and optimization of new materials with improved properties tailored for specific applications.
The goal of this short course is to provide an introduction to the theory and algorithms behind MD simulations, describe some of the most exciting recent developments in the field and exemplify with a few applications applications. The series also includes a tutorial on the nano-Materials Simulation Toolkit, an online MD simulation tool available at the nanoHUB. This provides users with a hands-on experience with MD simulations and enables further exploration of some of the concepts described in the lectures.
These lectures were taught at Purdue University during the Fall semester of 2008 as part of MSE597G “Modeling and Simulation of Materials”.
Course content
- An Introduction to Molecular Dynamics Unlimited
- Lecture 1: Classical Mechanics Unlimited
- Lecture 2: Statistical Mechanics I Unlimited
- Lecture 3: Statistical Mechanics II Unlimited
- Lecture 4: Interatomic potentials I Unlimited
- Lecture 5: Interatomic potentials II Unlimited
- Lecture 6: Interatomic potentials III Unlimited
- Lecture 7: Advanced Techniques for Molecular Dynamics Simulations Unlimited
N.A
- 5 stars0
- 4 stars0
- 3 stars0
- 2 stars0
- 1 stars0
No Reviews found for this course.



